

Efficient processes are critical for regulatory affairs teams aiming to bring new drugs to market. However, many organizations still depend on manual, decentralized tools like Excel and Outlook to navigate highly regulated and complex processes. While these tools may appear cost-effective and familiar, they pose practical challenges when applied to Investigational New Drug (IND) and New Drug Application (NDA) processes. These challenges can ultimately impede progress and lead to delays or regulatory consequences.
Discover the strategic pitfalls and practical obstacles inherent in manual regulatory affairs processes and uncover how automation holds the solution to unlocking growth, innovation, and compliance excellence in the life sciences industry.
High-level strategic risks of using manual tools for regulatory affairs processes
Clinging to outdated manual tools for facilitating regulatory affairs processes carries several profound strategic risks:
1. Missed opportunities
IND and NDA submissions are time-sensitive due to the intricacies of drug development and regulatory processes. Using manual tools for data collection, formatting, and submission can lead to submission delays, which, in turn, can:
- Extend development timelines: Delays can push back crucial clinical trials, affecting the overall development schedule.
- Affect market competitiveness: Timely approvals are vital in gaining a competitive edge in the pharmaceutical industry.
- Impact patient access: Swift approvals mean quicker access to potentially life-saving drugs for patients.
- Result in missed regulatory deadlines: Regulatory agencies enforce strict review timelines; missing them can lead to approval delays.
- Shake investor confidence and funding: Delays can affect investor and stakeholder confidence, potentially impacting funding.
- Disrupt clinical trials: Delayed IND submission can disrupt the recruitment and conduct of clinical trials.
2. Data inaccuracy
Manual data entry and management are prone to errors, which can affect the quality and accuracy of submissions. Inaccurate data can lead to questions and requests for clarification from regulatory authorities, prolonging the approval process.
3. Compliance delays
Manual tools lack the structure and control needed for strict regulatory compliance.
For example, in the IND and NDA processes, specific documentation requirements must be met. IND documentation requirements may include comprehensive preclinical study reports, detailed manufacturing processes, and clinical study protocols, while NDA documentation requirements may encompass labeling information, clinical trial summaries, and rigorous quality control data.
These documents must adhere to specific formats, standards, and timelines outlined by regulatory authorities like the FDA to ensure compliance and facilitate the review process. Failure to meet these requirements precisely can lead to regulatory delays, setbacks, or fines.
Practical obstacles of using manual regulatory affairs process tools
Those strategic risks don't exist in a vacuum; they stem from the tangible issues faced during the daily grind. Let's explore how the practical, day-to-day obstacles inherent in manual process tools create and amplify these strategic risks.
1. Data disorganization
Excel and Outlook are not designed to manage complex regulatory affairs data comprehensively. Storing critical information across multiple spreadsheets, emails, and documents can lead to data disorganization and difficulties in retrieving essential data when needed.
2. Version control issues
Collaboration in Excel and Outlook often results in version control challenges. In the regulatory affairs context, multiple teams and stakeholders may need to review and contribute to documents. Manual tools make it difficult to track changes, leading to confusion and potential errors.
3. Lack of process visibility
Processes in the regulatory affairs department are intricate and involve coordination across various departments. Manual tools lack visibility features, making it challenging to track the progress of tasks, set reminders, and allocate responsibilities efficiently.
Hypothetical case study
Wondering how these strategic risks and practical obstacles tend to manifest within an actual research and development department? Let's walk through a hypothetical scenario involving a fictional pharmaceutical company working to bring a groundbreaking drug called "Dimoufus " to market.
Throughout the R&D phase, this Dimoufus team encountered many challenges during both the IND and NDA processes. These challenges were rooted in their reliance on manual tools like Excel and Outlook, which are ill-suited for the complex, time-sensitive world of drug development.
From the start, data disorganization emerged as a significant problem during the IND phase. With critical documents scattered across different spreadsheets, emails, and drives, the Dimoufus team struggled to compile preclinical study reports, manufacturing processes, and clinical study protocols in a timely manner, causing delays in submission preparation. The impacts of data disorganization escalated during the documentation-heavy NDA phase — during which a crucial clinical safety report was inadvertently overlooked and nearly delayed the FDA review.
Strategic consequences of data disorganization:
- Missed opportunities: The delays caused by data disorganization extended the development timelines for Dimoufus, impacting their market competitiveness and patient access to the drug. Missed regulatory deadlines and investor confidence were also affected, representing missed opportunities for the company.
- Data inaccuracy: Rushed work due to data disorganization increased the likelihood of errors, jeopardizing the quality and accuracy of Dimoufus' submissions and potentially leading to regulatory queries.
- Compliance delays: The frantic search for data consumed the Dimoufus team's time and resources, resulting in rushed work and potential errors. This, in turn, delayed the submission process, putting them at risk of missing regulatory deadlines and incurring fines.
Version control issues further complicated matters. Collaboration across teams was vital, but shared spreadsheets and email attachments led to version control chaos. Revisions were made simultaneously, making it difficult to track changes accurately. This problem worsened during the NDA phase. Multiple versions of essential documents circulated, causing confusion and miscommunication among teams. Critical updates were inadvertently overwritten by outdated versions, jeopardizing the NDA submission.
Strategic consequences of version control issues:
- Missed opportunities: Version control issues led to inefficiencies in the review process, consuming valuable time and leaving less time for substantive review and improvement of the submission package. This could have affected the company's market competitiveness and patient access to the drug.
- Data inaccuracy: Overwriting critical updates with outdated versions posed a significant risk of data inaccuracy in Dimoufus' submissions, potentially resulting in regulatory queries and approval delays.
- Compliance delays: Version control challenges resulted in confusion and potential errors, which could have led to inaccuracies in Dimoufus' submissions. Regulatory authorities may require clarification, causing delays in the approval process.
The lack of process visibility compounded these challenges. During the IND process, the absence of real-time progress tracking made it impossible to monitor task responsibilities and deadlines. Delays became inevitable, undermining the drug development timeline. In the NDA phase, the complexity of tasks involving different departments worsened the problem. Without transparency, the Regulatory Affairs Project Manager struggled to allocate responsibilities and ensure timely task completion. Manual reminders and follow-ups added to the inefficiency.
Strategic consequences of poor process visibility:
- Compliance delays: The absence of real-time progress tracking made it challenging to monitor task responsibilities and deadlines, leading to delays in Dimoufus' drug development timeline. These delays could have resulted in missed regulatory deadlines.
- Missed opportunities: The lack of process visibility caused bottlenecks, miscommunications, and missed deadlines, which could have impacted Dimoufus' market competitiveness, patient access, and investor confidence. Delays could also disrupt clinical trials.
- Data inaccuracy: Inefficiencies caused by the lack of process visibility could lead to rushed work, increasing the potential for data inaccuracies in Dimoufus' submissions, which might prompt regulatory queries.
How automated solutions solve regulatory process headaches
As we've seen through the challenges faced by the Dimoufus team in our hypothetical case study, manual tools can create significant complications and stressors during crucial regulatory affairs processes. However, there's a way to streamline and simplify these processes, eliminating the need for scattered documents, version control woes, and visibility gaps. Enter automated solutions like Approvia.
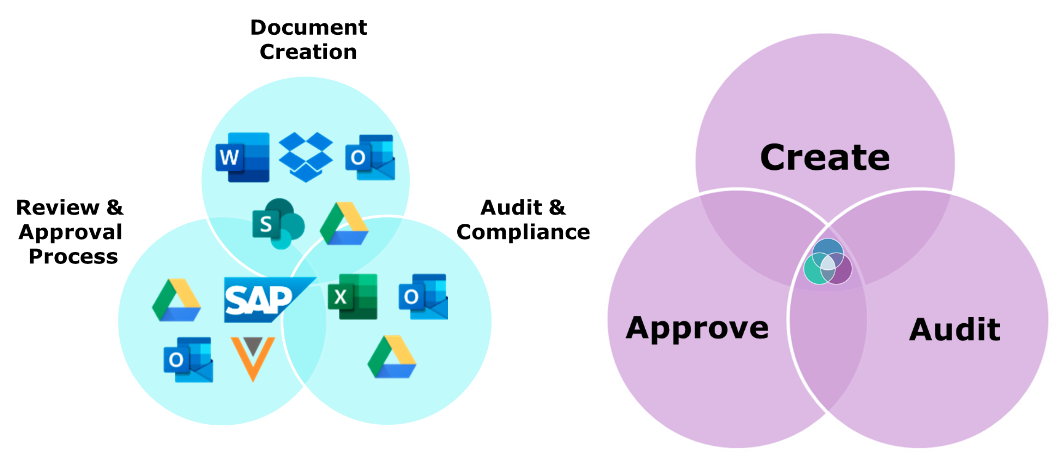
Approvia helps regulatory affairs teams take control of complex document management, bringing together critical functions like document creation, review and approval, and audit and compliance.
By centralizing regulatory document management, Approvia eliminates the need for password chasing, enhances collaboration with external stakeholders, and significantly reduces errors and compliance risks through comprehensive audit logs.
This powerful workflow automation tool offers a user-friendly experience, complete configurability to match your specific procedures, and affordability that sets it apart in the industry. With Approvia, you can structure your FDA submissions as dossiers, customize approval processes, and ensure seamless compliance with FDA guidelines—all in one cohesive platform.
How exactly does Approvia alleviate the strategic risks and tactical obstacles associated with manual tools like Excel and Outlook?
1. Compliance management
Approvia ensures regulatory compliance by providing structured templates for IND and NDA submissions, automatically formatting documents to meet FDA standards. It tracks compliance throughout the process, reducing the risk of compliance-related delays or fines.
2. Efficient data handling
Approvia centralizes data storage and management, eliminating the need for scattered spreadsheets and documents. It offers controlled access to authorized personnel, reducing data disorganization and streamlining data retrieval.
3. Version control and collaboration
Approvia offers collaborative document editing with version control. Teams can work on the same documents simultaneously, with changes tracked and easily reversible. This prevents version conflicts and ensures document accuracy.
4. Task and process visibility
Approvia provides a dashboard that offers a clear overview of the entire IND and NDA process. Users can set reminders, track task progress, and allocate responsibilities efficiently, enhancing process visibility and control.
5. Automated submission
Approvia simplifies the final submission process by compiling all approved sections into the required eCTD format. It conducts quality checks to ensure error-free submissions and offers integration with the Electronic Submissions Gateway for seamless FDA submissions.
From Excel to excellence
By harnessing the capabilities of specialized software solutions like Approvia, regulatory affairs departments within life sciences organizations can not only streamline their regulatory processes but also enhance compliance, accelerate drug development timelines, and ultimately contribute to the delivery of safe and effective treatments to patients.


